Multimaterial Thermoset Synthesis: Switching Polymerization Mechanism with Light Dosage

Ma, Y.; Drilling, R. J.; Recker, E. A.; Kim, J.-W.; Shankel, S. L.; Hu, J.; Easley, A. D.; Page, Z. A.; Lambert, T. H.; Fors, B. P. ACS Cent. Sci. 2024, ASAP.
The synthesis of polymeric thermoset materials with spatially controlled physical properties using readily available resins is a grand challenge. To address this challenge, we developed a photoinitiated polymerization method that enables the spatial switching of radical and cationic polymerizations by controlling the dosage of monochromatic light. This method, which we call Switching Polymerizations by Light Titration (SPLiT), leverages the use of substoichiometric amounts of a photobuffer in combination with traditional photoacid generators. Upon exposure to a low dose of light, the photobuffer inhibits the cationic polymerization, while radical polymerization is initiated. With an increased light dosage, the buffer system saturates, leading to the formation of a strong acid that initiates a cationic polymerization of the dormant monomer. Applying this strategy, patterning is achieved by spatially varying light dosage via irradiation time or intensity allowing for simple construction of multimaterial thermosets. Importantly, by the addition of an inexpensive photobuffer, such as tetrabutylammonium chloride, commercially available resins can be implemented in grayscale vat photopolymerization 3D printing to prepare sophisticated multimodulus constructs.
Electrophotocatalysis for Organic Synthesis

Lamb, M. C.; Steiniger, K. A.; Trigoura, L. K.; Wu, J.; Kundu, G.; Huang, H.; Lambert, T. H. Chem. Rev. 2024, 124, 122264-12304.
Electrocatalysis and photocatalysis have been the focus of extensive research efforts in organic synthesis in recent decades, and these powerful strategies have provided a wealth of new methods to construct complex molecules. Despite these intense efforts, only recently has there been a significant focus on the combined use of these two modalities. Nevertheless, the past five years have witnessed rapidly growing interest in the area of electrophotocatalysis. This hybrid strategy capitalizes on the enormous benefits of using photons as reagents while also employing an electric potential as a convenient and tunable source or sink of electrons. Research on this topic has led to a number of methods for C–H functionalization, reductive cross-coupling, and olefin addition among others. This field has also seen the use of a broad range of catalyst types, including both metal and organocatalysts. Of particular note has been work with open-shell photocatalysts, which tend to have comparatively large redox potentials. Electrochemistry provides a convenient means to generate such species, making electrophotocatalysis particularly amenable to this intriguing class of redox catalyst. This review surveys methods in the area of electrophotocatalysis as applied to organic synthesis, organized broadly into oxidative, reductive, and redox neutral transformations.
Hydrazine-Catalyzed Ring-Opening Metathesis Polymerization of Cyclobutenes.

Kellner-Rogers, J. S.; Hsu, J. H.; Keresztes, I.; Fors, B. P.; Lambert, T. H. Angew. Chem. Int. Ed. 2024, 63, e202413093. ChemRxiv. 10.26434/chemrxiv-2024-0qlff
Materials formed by the ring-opening metathesis polymerization (ROMP) of cyclic olefins are highly valued for industrial and academic applications but are difficult to prepare free of metal contaminants. Here we describe a highly efficient metal-free ROMP of cyclobutenes using hydrazine catalysis. Reactions can be initiated via in situ condensation of a [2.2.2]-bicyclic hydrazine catalyst with an aliphatic or aromatic aldehyde initiator. The polymerizations show living characteristics, achieving excellent control over molecular weight, low dispersity values, and high chain-end fidelity. Additionally, the hydrazine can be used in substoichiometric amounts relative to the aldehyde chain-end while maintaining good control over molecular weight and low dispersity values, indicating that a highly efficient chain transfer mechanism is occurring.
Accelerating Cationic Polymerizations with a Hydrogen Bond Donor

Shankel, S. L.; Ma, Y.; Spivey, J. A.; Filien, L.; Lambert, T. H.; Fors, B. P. Eur. Polym. J. 2024, 207, 112814.
Photoacid generators (PAGs) have facilitated a number of technology breakthroughs in the electronic, coating, and additive manufacturing industries. Traditionally, PAGs that contain weakly coordinating anions, such as PF6−, generate Brønsted superacids under UV irradiation for rapid cationic polymerizations. However, PAGs with strongly coordinating anions remain under-utilized as they form weak acids that are inefficient or even incapable of initiating polymerization. To expand the scope of potential counteranions in PAGs, we leveraged a thiophosphoramide hydrogen bond donor (HBD) to catalyze photoinitiated cationic polymerizations from diphenyliodonium PAGs. Through the formation of hydrogen bonds between the HBD and PAG counteranion, acceleration of the polymerization rate was observed for a range of non-coordinating and coordinating anions. The effect of the HBD on the polymerization kinetics was investigated by 1H − NMR titrations and geometry optimizations. Extending HBD catalysis beyond photopolymerizations, addition of HBD also enabled hydrochloric acid to initiate controlled reversible addition − fragmentation chain transfer (RAFT) polymerization under ambient conditions. With the versatility of HBD, there is potential to access initiation systems that were previously believed to be impractical for cationic polymerization.
Defect-Engineered Metal-Organic Frameworks as Bioinspired Heterogeneous Catalysts for Amide Bond Formation

Ahmad, B. I. Z.; Jerozal, R. T.; Meng, S.; Lambert, T. H.; Milner, P. J. ChemRxiv: 10.26434/chemrxiv-2023-65k97
The synthesis of amides from amines and carboxylic acids is the most widely carried out reaction in medicinal chemistry. Yet, most amide couplings are still carried out using stoichiometric reagents, leading to significant waste; few synthetic catalysts for this transformation have been adopted industrially due to their limited scope and/or poor recyclability. The majority of catalytic approaches focus on a single activation mode, such as enhancing the electrophilicity of the carboxylic acid partner using a Lewis acid. In contrast, nature effortlessly forges and breaks amide bonds using precise arrays of Lewis/Brønsted acidic and basic functional groups. Drawing inspiration from these systems, herein we report a simple defect engineering strategy to co-localize Lewis acidic Zr sites with other catalytically active species within porous metal-organic frameworks (MOFs). Specifically, the combination of pyridine N-oxide and Zr open metal sites within the framework MOF-808 produces a heterogeneous catalyst that facilitates amide bond formation with broad functional group compatibility. We propose that the formation of a hydrogen-bonding network at the defect sites helps to lower the energy barrier for amide bond formation. The defective MOF-808 catalyst can be recycled at least five times without losing crystallinity or catalytic activity. This defect engineering strategy can be potentially generalized to produce libraries of catalytically active MOFs with different combinations of co-localized active sites, mimicking the complexity of enzyme active sites.
Diazene-Catalyzed Oxidative Alkyl Halide-Olefin Metathesis

Kellner-Rogers, J. S.; Wang, R.; Lambert, T. H. Org. Lett. 2024, 26, 1078-1082. ChemRxiv: 10.26434/chemrxiv-2023-v1rbg
The first platform for oxidative alkyl halide-olefin metathesis is described. The procedure employs diazenes as catalysts, which effect cyclization of alkenyl alkyl halides to generate cyclic olefins and carbonyl products. The synthesis of phenanthrene, coumarin, and quinolone derivatives is demonstrated, as well as the potential to apply this strategy to other electrophiles.
Electrochemical Vicinal C–H Difunctionalization of Saturated Azaheterocycles
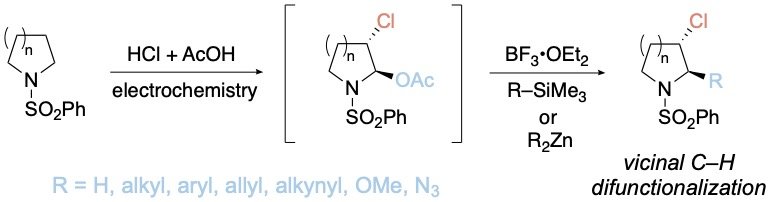
Kundu, G.; Lambert, T. H. J. Am. Chem. Soc. 2024, 146, 1794-1798.
A method to functionalize two vicinal C–H bonds of saturated azaheterocycles is described. The procedure involves subjecting the substrate to a mixture of hydrochloric acid, acetic acid, and acetic anhydride in an undivided electrochemical cell at a constant current, resulting in stereoselective conversion to the corresponding α-acetoxy-β-chloro derivative. The α-position can be readily substituted with a range of other groups, including alkyl, aryl, allyl, alkynyl, alkoxy, or azido functionalities. Furthermore, we demonstrate that the β-chloro position can be engaged in Suzuki cross-coupling. This protocol thus enables the rapid diversification of simple five-, six-, and seven-membered saturated azaheterocycles at two adjacent positions.
Cross-coupling of Amines via Photocatalytic Denitrogenation of In Situ-Generated Diazenes

Steiniger, K. A.; Lamb, M. C.; Lambert, T. H. J. Am. Chem. Soc. 2023, 145, 11524-11529.
A method for C(sp3)–C(sp3) cross-coupling of amines is described. Primary amines are converted to 1,2-dialkyldiazenes by treatment with O-nosylhydroxylamines in the presence of atmospheric oxygen. Denitrogenation of the diazenes with an iridium photocatalyst then forges the C–C bond. The substrate scope accommodates a broad latitude of functionality, including heteroaromatics and unprotected alcohols and acids.
Olefination of Carbonyls with Alkenes Enabled by Electrophotocatalytic Generation of Distonic Radical Cations

Steiniger, K. A.; Lambert, T. H. Science Adv. 2023, 9, eadg3026.
The conversion of carbonyls to olefins is a transformation of great importance for complex molecule synthesis. Standard methods use stoichiometric reagents that have poor atom economy and require strongly basic conditions, which limit their functional group compatibility. An ideal solution would be to catalytically olefinate carbonyls under nonbasic conditions using simple and widely available alkenes, yet no such broadly applicable reaction is known. Here, we demonstrate a tandem electrochemical/electrophotocatalytic reaction to olefinate aldehydes and ketones with a broad range of unactivated alkenes. This method involves the oxidation-induced denitrogenation of cyclic diazenes to form 1,3-distonic radical cations that rearrange to yield the olefin products. This olefination reaction is enabled by an electrophotocatalyst that inhibits back-electron transfer to the radical cation intermediate, thus allowing for the selective formation of olefin products. The method is compatible with a wide range of aldehydes, ketones, and alkene partners.
Electrophotocatalytic oxygenation of multiple adjacent C–H bonds

Shen, T.; Li, Y.-L.; Ye, K.-Y.; Lambert, T. H. Nature 2023, 614, 275-280.
Oxygen-containing functional groups are nearly ubiquitous in complex small molecules. The installation of multiple C–O bonds by the concurrent oxygenation of contiguous C–H bonds in a selective fashion would be highly desirable but has largely been the purview of biosynthesis. Multiple, concurrent C–H bond oxygenation reactions by synthetic means presents a challenge, particularly because of the risk of overoxidation. Here we report the selective oxygenation of two or three contiguous C–H bonds by dehydrogenation and oxygenation, enabling the conversion of simple alkylarenes or trifluoroacetamides to their corresponding di- or triacetoxylates. The method achieves such transformations by the repeated operation of a potent oxidative catalyst, but under conditions that are sufficiently selective to avoid destructive overoxidation. These reactions are achieved using electrophotocatalysis, a process that harnesses the energy of both light and electricity to promote chemical reactions. Notably, the judicious choice of acid allows for the selective synthesis of either di- or trioxygenated products.
Regiodivergent electrophotocatalytic aminooxygenation of aryl olefins.

Huang, H.; Lambert, T. H. J. Am. Chem. Soc. 2022, 144, 18803-18809.
A method for the regiodivergent aminooxygenation of aryl olefins under electrophotocatalytic conditions is described. The procedure employs a trisaminocyclopropenium (TAC) ion catalyst with visible light irradiation under a controlled electrochemical potential to convert aryl olefins to the corresponding oxazolines with high chemo- and diastereoselectivity. With the judicious choice between inexpensive and abundant reagents, namely water or urethane, either 2-amino-1-ol or 1-amino-2-ol derivatives could be prepared from the same substrate. This method is amenable to multigram synthesis of the oxazoline products with low catalyst loadings.
Interfacial electric fields catalyze Ullmann coupling reactions on gold surfaces.

Stone, I. B.; Starr, R. L.; Hoffmann, N.; Wang, X.; Evans, A. M.; Nuckolls, C.; Lambert, T. H.; Steigerwald, M. L.; Berkelbach, T. C.; Roy, X.; Venkataraman, L. Chem. Sci. 2022, 13, 10798-10805.
The electric fields created at solid–liquid interfaces are important in heterogeneous catalysis. Here we describe the Ullmann coupling of aryl iodides on rough gold surfaces, which we monitor in situ using the scanning tunneling microscope-based break junction (STM-BJ) and ex situ using mass spectrometry and fluorescence spectroscopy. We find that this Ullmann coupling reaction occurs only on rough gold surfaces in polar solvents, the latter of which implicates interfacial electric fields. These experimental observations are supported by density functional theory calculations that elucidate the roles of surface roughness and local electric fields on the reaction. More broadly, this touchstone study offers a facile method to access and probe in real time an increasingly prominent yet incompletely understood mode of catalysis.
Cyclopropenium Ions in Catalysis.

Wilson, R. M.; Lambert, T. H. Acc. Chem. Res. 2022, 55, 3057-3069.
Cyclopropenium ions are the smallest class of aromatic compounds, satisfying Hückel’s rules of aromaticity with two π electrons within a three-membered ring. First prepared by Breslow in 1957, cyclopropenium ions have been found to possess extraordinary stability despite being both cationic and highly strained. In the 65 years since their first preparation, cyclopropenium ions have been the subject of innumerable studies concerning their synthesis, physical properties, and reactivity. However, prior to our work, the reactivity of these unique carbocations had not been exploited for reaction promotion or catalysis.
Over the past 13 years, we have been exploring aromatic ions as unique and versatile building blocks for the development of catalysts for organic chemistry. A major portion of this work has been focused on leveraging the remarkable properties of the smallest of the aromatic ions─cyclopropeniums─as a design element in the invention of highly reactive catalysts. Indeed, because of its unique profile of hydrolytic stability, compact geometry, and relatively easy oxidizability, the cyclopropenium ring has proven to be a highly advantageous construction module for catalyst invention.
In this Account, we describe some of our work using cyclopropenium ions as a key element in the design of novel catalysts. First, we discuss our early work aimed at promoting dehydrative reactions, starting with Appel-type chlorodehydrations of alcohols and carboxylic acids, cyclic ether formations, and Beckmann rearrangements and culminating in the realization of catalytic chlorodehydrations of alcohols and a catalytic Mitsunobu-type reaction. Next, we describe the development of cyclopropenimines as strong, neutral organic Brønsted bases and, in particular, the use of chiral cyclopropenimines for enantioselective Brønsted catalysis. We also describe the development of higher-order cyclopropenimine superbases. The use of tris(amino)cyclopropenium (TAC) ions as a novel class of phase-transfer catalysts is discussed for the reaction of epoxides with carbon dioxide. Next, we describe the formation of a cyclopropenone radical cation that has a portion of its spin density on the oxygen atom, leading to some peculiar metal ligand behavior. Finally, we discuss recent work that employs TAC electrophotocatalysts for oxidation reactions. The key intermediate for this chemistry is a TAC radical dication, which as an open-shell photocatalyst has remarkably strong excited-state oxidizing power. We describe the application of this strategy to transformations ranging from the oxidative functionalization of unactivated arenes to the regioselective derivatization of ethers, C–H aminations, vicinal C–H diaminations, and finally aryl olefin dioxygenations. Collectively, these catalytic platforms demonstrate the utility of charged aromatic rings, and cyclopropenium ions in particular, to enable unique advances in catalysis.
Highly Twisted Azobenzene Ligand Causes Crystals to Continuously Roll in Sunlight.

Bartholomew, A. K.; Stone, I. B.; Steigerwald, M. L.; Lambert, T. H.; Roy, X. J. Am. Chem. Soc. 2022, 144, 16773-16777.
Direct conversion of solar energy to mechanical work promises higher efficiency than multistep processes, adding a key tool to the arsenal of energy solutions necessary for our global future. The ideal photomechanical material would convert sunlight into mechanical motion rapidly, without attrition, and proportionally to the stimulus. We describe crystals of a tetrahedral isocyanoazobenzene–copper complex that roll continuously when irradiated with broad spectrum white light, including sunlight. The rolling results from bending and straightening of the crystal due to blue light-driven isomerization of a highly twisted azobenzene ligand. These findings introduce geometrically constrained crystal packing as a strategy for manipulating the electronic properties of chromophores. Furthermore, the continuous, solar-driven motion of the crystals demonstrates direct conversion of solar energy to continuous physical motion using easily accessed molecular systems.
Moisture tolerant cationic RAFT polymerization of vinyl ethers

Shankel, S. L.; Lambert, T. H.; Fors, B. P. Polymer Chem. 2022, 13, 5974-5979.
Cationic reversible addition–fragmentation chain transfer (RAFT) polymerizations have permitted the controlled polymerization of vinyl ethers and select styrenics with predictable molar masses and easily modified thiocarbonylthio chain ends. However, most cationic RAFT systems require inert reaction conditions with highly purified reagents and low temperatures. Our groups recently developed a living cationic polymerization that does not require these rigorous conditions by utilizing a strong organic acid (pentacarbomethoxycyclopentadiene (PCCP)) and a hydrogen bond donor. By combining our PCCP acid promoted polymerization with a chain transfer agent, we have designed a tolerant cationic RAFT system that can be performed neat, open to the air, and at room temperature. Additionally, this system allows us to utilize catalytic amounts of the PCCP acid to furnish polymers with chain end functionality that can be easily isolated and further manipulated to make functional materials.
Poly(2,3-Dihydrofuran): A Strong, Biorenewable, and Degradable Thermoplastic Synthesized via Room Temperature Cationic Polymerization

Spring, S.; Hsu, J.; Sifri, R.; Yang, S.-M.; Cerione, C.; Lambert, T. H.; Ellison, C.; Fors, B. J. Am. Chem. Soc. 2022, 144, 15727-15734.
Creation of strong and tough plastics from sustainable and biorenewable resources is a significant challenge in polymer science. This challenge is further complicated when attempting to make these materials using an economically viable process, which is often hindered by the production and availability of chemical feedstocks and the efficiency of the monomer synthesis. Herein, we report the synthesis and characterization of a strong thermoplastic made from 2,3-dihydrofuran (DHF), a monomer made in one step from 1,4-butanediol, a bioalcohol already produced on the plant scale. We developed a green, metal-free cationic polymerization to enable the production of poly(2,3-dihydrofuran) (PDHF) with molecular weights of up to 256 kg/mol at room temperature. Characterization of these polymers showed that PDHF possesses high tensile strength and toughness (70 and 14 MPa, respectively) comparable to commercial polycarbonate, high optical clarity, and good barrier properties to oxygen, carbon dioxide, and water. These properties make this material amenable to a variety of applications, from food packaging to high strength windows. Importantly, we have also developed a facile oxidative degradation process of PDHF, providing an end-of-life solution for PDHF materials.
Electrophotocatalysis: Combining Light and Electricity to Catalyze Reactions

Huang, H.; Steiniger, K. A.; Lambert, T. H. J. Am. Chem. Soc. 2022, 144, 12567-12583.
Visible-light photocatalysis and electrocatalysis are two powerful strategies for the promotion of chemical reactions that have received tremendous attention in recent years. In contrast, processes that combine these two modalities, an area termed electrophotocatalysis, have until recently remained quite rare. However, over the past several years a number of reports in this area have shown the potential of combining the power of light and electrical energy to realize new catalytic transformations. Electrophotocatalysis offers the ability to perform photoredox reactions without the need for large quantities of stoichiometric or superstoichiometric chemical oxidants or reductants by making use of an electrochemical potential as the electron source or sink. In addition, electrophotocatalysis is readily amenable to the generation of open-shell photocatalysts, which tend to have exceptionally strong redox potentials. In this way, potent yet selective redox reactions have been realized under relatively mild conditions. This Perspective highlights recent advances in the area of electrophotocatalysis and provides some possible avenues for future work in this growing area.
Metal–Free Ring–Opening Metathesis Polymerization with Hydrazonium Initiators

Quach, P.; Hsu, J. H.; Keresztes, I.; Fors, B. P.; Lambert, T. H. Angew. Chem. Int. Ed. 2022, 61 , e202203344. Preprint: 10.26434/chemrxiv-2022-q8n0b
A new strategy for the ring–opening metathesis polymerization (ROMP) of cycloalkenes using hydrazonium initiators is described. The initiators, which are formed by the condensation of 2,3-diazabicyclo[2.2.2]octane and an aldehyde, polymerize cyclopropene monomers by a sequence of [3+2] cycloaddition and cycloreversion reactions. This process generates short chain polyolefins (Mn ≤ 9.4 kg/mol) with relatively low dispersities (Đ ≤ 1.4). The optimized conditions showed efficiency comparable to that achieved with Grubbs’ catalyst. A positive correlation between monomer to initiator ratio and degree of polymerization was revealed through NMR spectroscopy.
Polyimide as a Durable Cathode for All-Solid-State Li(Na)-Organic Batteries with Boosted Cell-Level Energy Density.

Ji, W.; Zhang, X.; Qu, H.; Xin, L.; Luedtke, A.; Huang, H.; Lambert, T. H.; Qu, D. Nano Energy 2022, 96, 107130.
The integration of organic electrode materials (OEMs) with solid-state electrolytes (SSEs) is expected to build an all-solid-state battery (ASSB) with long-term sustainability, high safety, and high energy density. Despite this great promise, the cell-level energy density is still far from practically applicable, which stems from the ultrathick SSE layer and thin cathode layer used in a pellet-type ASSB design. Here, a cost-effective polyimide (PI) material was first exploited as an organic cathode for sulfide-based ASSBs. A capacity of ~190 mAh g−1 was delivered with almost no capacity decay over 300 cycles. Moreover, for the first time, a dry-film approach was introduced to manufacture a sheet-type Li−organic ASSB with an ultrathin SSE layer and a high-areal-loading PI cathode. Notably, PI is a perfect candidate for dry-film technology due to its high thermal stability and extraordinary chemical inertness toward sulfide SSEs. Remarkably, the free-standing SSE membrane was merely 46 µm thick, and an ultralow areal resistance of 3.3 Ω cm2 was achieved, more than tenfold lower than that of reported SSE pellets. One order of magnitude boost in the cell-level energy density was achieved. This work presents a significant leap in transferring organic ASSB technology from laboratory research to factory manufacturing.
Practically Accessible All-Solid-State Batteries Enabled by Organosulfide Cathodes and Sulfide Electrodes
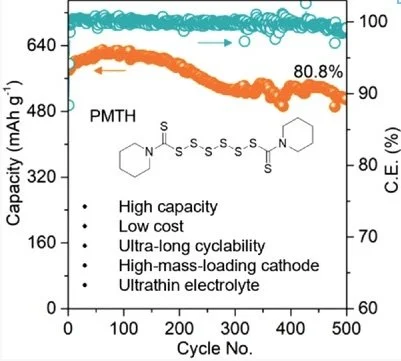
Ji, W.; Zhang, X.; Zheng, D.; Huang, H.; Lambert, T. H.; Qu, D. Adv. Funct. Mater. 2022, 2202919.
The combination of organic electrode materials and sulfide electrolytes is expected to enable the development of all-solid-state organic batteries featuring high energy density, safety, and sustainability. Here, thiuram hexasulfide is first reported as a low-cost and high-capacity cathode material for solid-state organic batteries based on sulfide electrolytes. Notably, a capacity of ≈600 mA h g−1 is delivered and the capacity retention is 80.8% after 500 cycles. An electrochemically reversible change of the cathode interface is revealed upon cycling. The full cell displays an oscillating stress change of up to 0.6 MPa during cycling, predominated by the anode side. The energy density is 1140 Wh kg−1 at the material level and 376 Wh kg−1 at the electrode level, which are among the best-reported organic cathodes to date. A high areal capacity of 10.4 mA h cm−2 is reached with a high mass loading cathode. A dry-film approach is further explored to manufacture sheet-type cells. The free-standing Li6PS5Cl film with a thickness of only 48 µm demonstrates an ultralow areal resistance of 3.9 Ω cm2, which significantly boosts the cell-level energy density and reduces the cell internal resistance.
Ring-Opening Carbonyl-Olefin Metathesis of Cyclobutenes
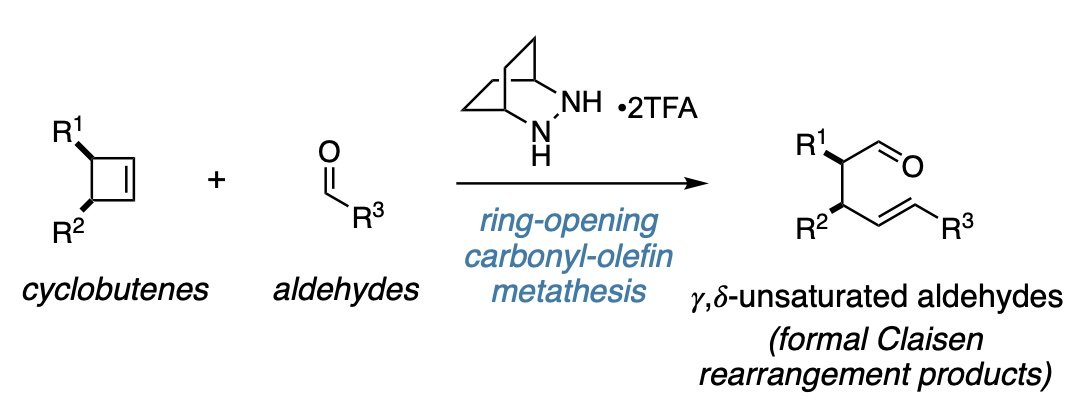
Holl, M. G.; Lambert, T. H. ACS Catal. 2022, 12, 4813-4817.
The ring-opening carbonyl-olefin metathesis of cyclobutenes to furnish gamma, delta-unsaturated aldehydes—formal Claisen rearrangement products—is reported. The bistrifluoroacetic acid salt of 2,3-diazabicyclo[2.2.2]octane promotes these reactions efficiently with a variety of cyclobutenes and aldehydes, including aliphatic, alpha, beta-unsaturated, aryl, and heteroaryl aldehydes. Catalytic reactions are also demonstrated.
Polycyclic Heteroaromatics via Hydrazine-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis

Cho, E. K.; Quach, K. P.; Zhang, Y.; Sim, J. H.; Lambert, T. H. Chem. Sci. 2022, 13, 2418-2422.
The use of hydrazine-catalyzed ring-closing carbonyl-olefin metathesis (RCCOM) to synthesize polycyclic heteroaromatic (PHA) compounds is described. In particular, substrates bearing Lewis basic functionalities such as pyridine rings and amines, which strongly inhibit acid catalyzed RCCOM reactions, are shown to be compatible with this reaction. Using 5 mol% catalyst loadings, a variety of PHA structures can be synthesized in from biaryl alkenyl aldehydes, which themselves are readily prepared by cross-coupling.
A High-Performance Organic Cathode Customized for Sulfide-based All-solid-state Batteries

Ji, W.; Zhang, X.; Xin, L.; Luedtke, A.; Zheng, D.; Huang, H.; Lambert, T. H.; Qu, D. Energy Stor. Mater. 2022, 45, 680-686.
All-solid-state batteries (ASSBs) have become increasingly attractive recently due to their better safety and prospective long-term stability compared with conventional liquid batteries. However, obtaining a sustainable cathode candidate to match the solid electrolyte with regards to operating potential, chemical compatibility, and mechanical property is still an open challenge. Herein, the chemical incompatibility of quinone-based active materials and sulfide-based electrolyte were unveiled for the first time through a heteroconjugate addition reaction mechanism. To develop a quinone cathode customized for sulfide-based ASSBs, poly-(anthraquinonyl sulfide)-graphene (PAQS-G) nanocomposite was reported. The stable polymer framework of PAQS can protect the quinone redox center by preventing nucleophilic attack from sulfide-based solid electrolytes. The graphene additives can ameliorate redox kinetics and improve active material utilization. The PAQS-G cathode exhibited a specific capacity of ∼178 mAh g−1 and a high material utilization of ∼79%. Excellent cycling stability was achieved with 94 % capacity after 200 cycles in lithium batteries and 95.5 % capacity after 300 cycles in sodium batteries at 0.1C rate, respectively. A promising potential for energy storage applications was demonstrated.
Primary Alcohols via Nickel Pentacarboxycyclopentadienyl Diamide–Catalyzed Hydrosilylation of Terminal Epoxides

Steiniger, K. A.; Lambert, T. H. Org. Lett. 2021, 23, 8013-8017.
The efficient and regioselective hydrosilylation of epoxides co-catalyzed by a pentacarboxycyclopentadienyl (PCCP) diamide nickel complex and Lewis acid is reported. This method allows for the reductive opening of terminal, monosubstituted epoxides to form unbranched, primary alcohols. A range of substrates including both terminal and non-terminal epoxides are shown to work, and a mechanistic rationale is provided. This work represents the first use of a PCCP derivative as a ligand for transition metal catalysis.
A Single-Molecule Blueprint for Synthesis

Stone, I.; Starr, R. L.; Zang, Y.; Nuckolls, C.; Steigerwald, M. L.; Lambert, T. H.; Roy, X.; Venkataraman, L. Nat. Rev. Chem. 2021, 5, 695-710.
Chemical reactions that occur at nanostructured electrodes have garnered widespread interest because of their potential applications in fields including nanotechnology, green chemistry and fundamental physical organic chemistry. Much of our present understanding of these reactions comes from probes that interrogate ensembles of molecules undergoing various stages of the transformation concurrently. Exquisite control over single-molecule reactivity lets us construct new molecules and further our understanding of nanoscale chemical phenomena. We can study single molecules using instruments such as the scanning tunnelling microscope, which can additionally be part of a mechanically controlled break junction. These are unique tools that can offer a high level of detail. They probe the electronic conductance of individual molecules and catalyse chemical reactions by establishing environments with reactive metal sites on nanoscale electrodes. This Review describes how chemical reactions involving bond cleavage and formation can be triggered at nanoscale electrodes and studied one molecule at a time.
Enantioenriched α-Substituted Glutamates / Pyroglutamates via Enantioselective Cyclopropenimine-Catalyzed Michael Addition of Amino Ester Imines

Seibel, Z. M.; Bandar, J. S.; Lambert, T. H. Beilstein J. Org. Chem. 2021, 17, 2077-2084. *Invited contribution for special issue on asymmetric organocatalysis
A procedure for the enantioselective synthesis of a-substituted glutamates and pyroglutamates via cyclopropenimine-catalyzed Michael addition of amino ester imines is described. Enantioselectivities of up to 94% have been achieved, and a variety of functional groups were found to be compatible. The impact of catalyst structure and imine substitution is discussed. Compared to other methods, this protocol allows for broader and more enantioselective access to pyroglutamate derivatives.
Carbonyl-Olefin Metathesis

Albright, H.; Davis, A. J.; Gomez-Lopez, J. L.; Vonesh, H. L.; Quach, P. K.; Lambert, T. H.; Schindler, C. S. Chem. Rev. 2021, 121, 9359-9406.
This Review describes the development of strategies for carbonyl–olefin metathesis reactions relying on stepwise, stoichiometric, or catalytic approaches. A comprehensive overview of currently available methods is provided starting with Paternò–Büchi cycloadditions between carbonyls and alkenes, followed by fragmentation of the resulting oxetanes, metal alkylidene-mediated strategies, [3 + 2]-cycloaddition approaches with strained hydrazines as organocatalysts, Lewis acid-mediated and Lewis acid-catalyzed strategies relying on the formation of intermediate oxetanes, and protocols based on initial carbon–carbon bond formation between carbonyls and alkenes and subsequent Grob-fragmentations. The Review concludes with an overview of applications of these currently available methods for carbonyl–olefin metathesis in complex molecule synthesis. Over the past eight years, the field of carbonyl–olefin metathesis has grown significantly and expanded from stoichiometric reaction protocols to efficient catalytic strategies for ring-closing, ring-opening, and cross carbonyl–olefin metathesis. The aim of this Review is to capture the status quo of the field and is expected to contribute to further advancements in carbonyl–olefin metathesis in the coming years.
C−H Amination via Electrophotocatalytic Ritter-type Reaction
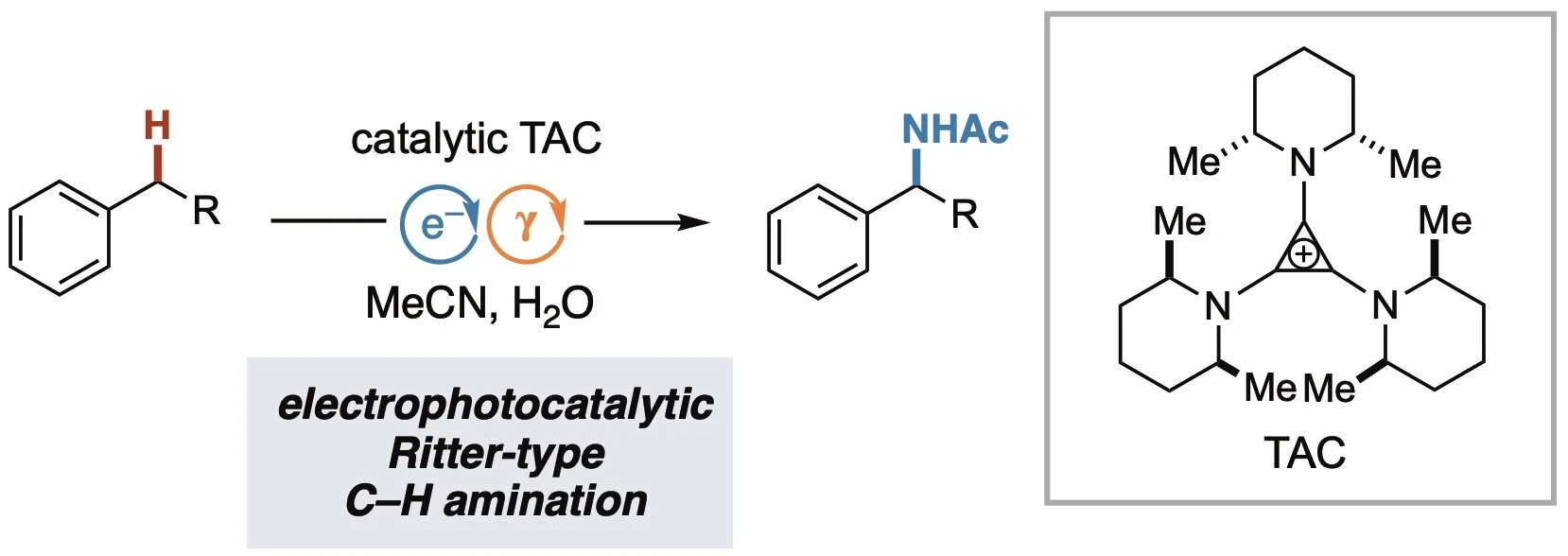
Shen, T.; Lambert, T. H. J. Am. Chem. Soc. 2021, 143, 8597-8602.
A method for C−H bond amination via an electrophotocatalytic Ritter-type reaction is described. The reaction is catalyzed by a trisaminocyclopropenium (TAC) ion in an electrochemical cell under irradiation. These conditions convert benzylic C−H bonds to acetamides without the use of a stoichiometric chemical oxidant. A range of functionality is shown to be compatible with this transformation, and several complex substrates are demonstrated.
Electrophotocatalytic Acetoxyhydroxylation of Aryl Olefins

Huang, H.; Lambert, T. H. J. Am. Chem. Soc. 2021, 143, 7247-7252.
A method for the acetoxyhydroxylation of olefins with syn stereoselectivity under electrophotocatalytic conditions is described. The procedure uses a trisaminocyclopropenium (TAC) ion catalyst with visible light irradiation under a controlled electrochemical potential to convert aryl olefins to the corresponding glycol monoesters with high chemo- and diastereoselectivity. This reaction can be performed in batch or in flow, enabling multigram synthesis of the monoester products.
Electrophotocatalytic C–H Heterofunctionalization of Arenes

Huang, H.; Lambert, T. H. Angew. Chem. Int. Ed. 2021, 60, 11163-11167.
The electrophotocatalytic heterofunctionalization of arenes is described. Using 2,3‐dichloro‐5,6‐dicyanoquinone (DDQ) under a mild electrochemical potential with visible light irradiation, arenes undergo oxidant‐free hydroxylation, alkoxylation, and amination with high chemoselectivity. In addition to batch reactions, an electrophotocatalytic recirculating flow process is demonstrated, enabling the conversion of benzene to phenol on a gram scale.
Electrophotocatalytic Diamination of Vicinal C–H Bonds.

Shen, T.; Lambert, T. H. Science 2021, 371, 620-626.
The conversion of unactivated carbon-hydrogen (C–H) bonds to carbon–nitrogen (C–N) bonds is a highly valued transformation. Existing strategies typically accomplish such reactions at only a single C–H site because the first derivatization diminishes the reactivity of surrounding C–H bonds. Here, we show that alkylated arenes can undergo vicinal C–H diamination reactions to form 1,2-diamine derivatives through an electrophotocatalytic strategy, using acetonitrile as both solvent and nitrogen source. The reaction is catalyzed by a trisaminocyclopropenium (TAC) ion, which undergoes anodic oxidation to furnish a stable radical dication while the cathodic reaction reduces protons to molecular hydrogen. Irradiation of the TAC radical dication (wavelength of maximum absorption of 450 to 550 nanometers) with a white-light compact fluorescent light generates a strongly oxidizing photoexcited intermediate.
Hydrogen Bond Donor Catalyzed Cationic Polymerization of Vinyl Ethers.

Kottisch, V.; Jermaks, J.; Mak, J.-Y.; Woltornist, R. A.; Lambert, T. H.; Fors, B. P. Angew. Chem. Int. Ed. 2021, 60, 4535-4539.
The synthesis of high‐molecular‐weight poly(vinyl ethers) under mild conditions is a significant challenge, since cationic polymerization reactions are highly sensitive to chain‐transfer and termination events. We identified a novel and highly effective hydrogen bond donor (HBD)–organic acid pair that can facilitate controlled cationic polymerization of vinyl ethers under ambient conditions with excellent monomer compatibility. Poly(vinyl ethers) of molar masses exceeding 50 kg mol−1 can be produced within 1 h without elaborate reagent purification. Modification of the HBD structure allowed tuning of the polymerization rate, while DFT calculations helped elucidate crucial intermolecular interactions between the HBD, organic acid, and polymer chain end.
A Redox-Active Organic Cation for Safer Metallic Lithium-Based Batteries.

Ji, W.; Huang, H.; Zheng, D.; Zhang, X.; Ding, T.; Lambert, T. H.; Qu, D. Energy Stor. Mater. 2020, 32, 185-190.
Safety concerns have severely impeded the practical application of high-energy-density lithium-based batteries. Dendrite growth and overcharging can lead to particularly catastrophic thermal failure. Here we report an organic cation, trisaminocyclopropenium (TAC), as a bi-functional electrolyte additive to suppress dendrite growth and offer reversible overcharge protection for metallic lithium-based batteries. During the Li plating process, TAC cations with aliphatic chains can form a positively charged electrostatic shield around Li protrusions, repelling the approaching Li+ and thereby attaining a more uniform plating. A two times longer cycle life of 300 h at 1 mA cm−2 is achieved in a Li|Li symmetric cell in comparison with the control. During the overcharging process, the redox-active TAC can repeatedly shuttle between two electrodes, maintaining the cell voltage within a safe value. A solid protection of 117 cycles (~1640 h) at 0.2 C with a 100% overcharge is achieved in a LiFePO4/Li4Ti5O12 cell. This study sheds fresh light on the ability of organic cations to build safer batteries.
A Redox-Active Organic Cation for Safer High Energy Density Li-Ion Batteries

Ji, W.; Huang, H.; Huang, X.; Zhang, X.; Zheng, D.; Ding, T.; Chen, J.; Lambert, T. H.; Qu, D. J. Mat. Chem. A 2020, 8, 17156-17162.
Ni-rich layered cathode materials are at the forefront to be deployed in the high energy density Li-ion batteries for the automotive market. However, the intrinsic poor structural and interfacial stability during overcharging could trigger violent thermal failure, which severely limits their wide application. To protect the Ni-rich cathode from overcharging, we firstly report a redox-active cation, thioether-substituted diaminocyclopropenium, as an electrolyte additive to limit the cell voltage within the safe value during overcharging. The organic cation demonstrates a record-breaking electrochemical reversibility at ~4.55 V versus Li+/Li and solubility (0.5 M) in carbonate-based electrolyte. The protection capability of the additive was explored in two cell chemistries: a LiNi0.8Co0.15Al0.05O2/graphite cell and a LiNi0.8Co0.15Al0.05O2/silicon-graphene cell with an areal capacity of ~2.2 mAh cm-2 and ~3 mAh cm-2, respectively. With 0.2 M addition, the LiNi0.8Co0.15Al0.05O2/graphite cell survived 54 cycles at 0.2 C with 100% overcharge. Moreover, the cell can carry an utmost 4.4 mA cm-2 (2 C) with 100% overcharge and a maximum capacity of 7540% SOC at 0.2 C.
Synthesis of 1,2-Dihydroquinolines via Hydrazine-Catalysed Ring-Closing Carbonyl-Olefin Metathesis.

Zhang, Y.; Sim, J. H.; MacMillan, S. N.; Lambert, T. H. Org. Lett. 2020, 22, 6026-6030. Preprint: ChemRxiv, https://doi.org/10.26434/chemrxiv.11342477.v1
The synthesis of 1,2-dihydroquinolines by the hydrazine-catalysed ring-closing carbonyl-olefin metathesis (RCCOM) of N-prenylated 2-aminobenzaldehydes is reported. Substrates with a variety of substitution patterns are shown, and the compatibility of these conditions with a range of additives is demonstrated. With an acid-labile protecting group on the nitrogen atom, in situ deprotection and autoxidation furnishes quinolines. In comparison to related oxygen-containing substrates, the cycloaddition step of the catalytic cycle is shown to be slower, but the cycloreversion is found to be more facile.
Ring-Opening Carbonyl-Olefin Metathesis of Norbornenes.

Jermaks, J.; Quach, P.; Seibel, Z. M.; Pomarole, J.; Lambert, T. H. Chem. Sci. 2020, 11, 7884-7895. Preprint: ChemRxiv, https://doi.org/10.26434/chemrxiv.11385774.v1
A computational and experimental study of the hydrazine-catalyzed ring-opening carbonyl-olefin metathesis of norbornenes is described. Detailed theoretical investigation of the energetic landscape for the full reaction pathway with six different hydrazines revealed several crucial aspects for the design of next-generation hydrazine catalysts. This study indicated that a [2.2.2]-bicyclic hydrazine should offer substantially increased reactivity versus the previously reported [2.2.1]-hydrazine due to a lowered activation barrier for the rate-determining cycloreversion step, a prediction which was verified experimentally. Optimized conditions for both cycloaddition and cycloreversion steps were identified, and a brief substrate scope study for each was conducted. A complication for catalysis was found to be the slow hydrolysis of the ring-opened hydrazonium intermediates, which were shown to suffer from a competitive and irreversible cycloaddition with a second equivalent of norbornene. This problem was overcome by the strategic incorporation of a bridgehead methyl group on the norbornene ring, leading to the first demonstrated catalytic carbonyl-olefin metathesis of norbornene rings.
A Redox-active Organic Salt for Safer Na-ion Batteries.

Ji, W.; Huang, H.; Zhang, X.; Zheng, D.; Ding, T.; Lambert, T. H.; Qu, D. Nano Energy, 2020, https://doi.org/10.1016/j.nanoen.2020.104705
Overcharge abuse can trigger thermal runaway when a device is left unattended. Redox shuttles, as economic and efficient electrolyte additives, have been proven to provide reliable and reversible protection for state-of-art Li-ion batteries (LIBs) against overcharge. Here, a functional organic salt, trisaminocyclopropenium perchlorate (TAC•ClO4), is developed and employed as a redox shuttle for overcharge protection in a Na-ion battery system. This type of novel redox shuttle molecule is reported for the first time. As a unique ionic compound with the smallest aromatic ring structure, TAC•ClO4 exhibits distinctive attributes of fast diffusion, high solubility, and ultrahigh chemical / electrochemical stability in both redox states. With merely 0.1 M TAC•ClO4 in electrolyte, Na3V2(PO4)3 cathode can carry overcharge current even up to 10C or 400% SOC. Na3V2(PO4)3/hard carbon cells demonstrated strong anti-overcharging ability of 176 cycles at 0.5C rate and 54 cycles at 1C rate with 100% overcharge. Moreover, TAC•ClO4 addition has little impact on the electrochemical performance of Na-ion batteries, especially on the rate performance and the initial Columbic efficiency. Interestingly, a unique and reversible electrochromic behavior of TAC•ClO4 electrolyte can promptly provide the device an overcharge alarm under a designed potential to further enhance the safety level.
Self-Assembly of Aminocyclopropenium Salts: En Route to Deltic Ionic Liquid Crystals.

Litterscheidt, J.; Bandar, J. S.; Ebert, M.; Forschner, R.; Bader, K.; Lambert, T. H.; Frey, W.; Bühlmeyer, A.; Brändle, M.; Schulz, F.; Laschat. S. Angew. Chem. Int. Ed. 2020, 59, 10557-10565.
Aminocyclopropenium ions have raised much attention both as organocatalysts as well as redoxactive polymers. The self‐assembly of amphiphilic aminocyclopropenium ions remains challenging however. Here the first deltic ionic liquid crystals based on aminocyclopropenium ions have been developed. Differential scanning calorimetry (DSC), polarizing optical microscopy (POM) and X‐ray diffraction (WAXS, SAXS) provided insight into the unique self‐assembly and nanosegregation of these liquid crystals. While the combination of small headgroups with linear p ‐alkoxyphenyl units led to bilayer‐type smectic mesophases, wedge‐shaped units resulted in columnar mesophases. Upon increasing the size and polyphilicity of the aminocyclopropenium headgroup again a lamellar phase was formed.
Reductive Electrophotocatalysis: Merging Electricity and Light to Achieve Extreme Reduction Potentials.

Kim, H.; Kim, H.; Lambert, T. H.; Lin, S. J. Am. Chem. Soc. 2020, 142, 2087-2092.
We describe a new electrophotocatalytic strategy that harnesses the power of light and electricity to generate an excited radical anion with a reducing potential of −3.2 V vs SCE, which can be used to activate substrates with very high reduction potentials (Ered ≈ −1.9 to −2.9 V). The resultant aryl radicals can be engaged in various synthetically useful transformations to furnish arylboronate, arylstannane, and biaryl products.
Electrophotocatalytic C–H Functionalization of Ethers with High Regioselectivity.

Huang, H.; Strater, Z. M.; Lambert, T. H. J. Am. Chem. Soc. 2020, 142, 1698-1703.
The highly regioselective electrophotocatalytic C–H functionalization of ethers is described. These reactions are catalyzed by a trisaminocyclopropenium (TAC) ion at mild electrochemical potential with visible light irradiation. Ethers undergo oxidant-free coupling with isoquinolines, alkenes, alkynes, pyrazoles, and purines with typically high regioselectivity for the less-hindered α-position. The reaction is proposed to operate via hydrogen atom transfer (HAT) from the substrate to the photoexcited TAC radical dication, thus demonstrating a new reactivity mode for this electrophotocatalyst.
Room Temperature, Base-Free SNAr Reactions with Unactivated Aryl Fluorides.

Huang, H.; Lambert, T. H. Angew. Chem. Int. Ed. 2020, 59, 658-662.
The electrophotocatalytic SNAr reaction of unactivated aryl fluorides at ambient temperature without strong base is demonstrated.
Synthesis of 2H-Chromenes via Hydrazine-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis.

Zhang, Y.; Jermaks, J.; MacMillan, S. N.; Lambert, T. H. ACS Catal. 2019, 9, 9259-9264.
The catalytic ring-closing carbonyl-olefin metathesis (RCCOM) of O-allyl salicylaldehydes to form 2H-chromenes is described. The method utilizes a [2.2.1]-bicyclic hydrazine catalyst and operates via a [3 + 2]/retro-[3 + 2] metathesis manifold. The nature of the allyl substitution pattern was found to be crucial, with sterically demanding groups such as adamantylidene or diethylidene offering optimal outcomes. A survey of substrate scope is shown along with a discussion of mechanism supported by DFT calculations. Steric pressure arising from syn-pentane minimization of the diethylidene moiety is proposed to facilitate cycloreversion.
In Situ Coupling of Single Molecules Driven by Au-Catalyzed Electrooxidation.

Zang, Y.; Stone, I.; Inkpen, M. S.; Liu, Z.-F.; Ng, F.; Lambert, T. H.; Nuckolls, C.; Steigerwald, M. L.; Roy, X.; Venkataraman, L. Angew. Chem. Int. Ed. 2019, 58, 16008-16012.
A single‐molecule method has been developed based on the scanning tunneling microscope (STM) to selectively couple a series of aniline derivatives and create azobenzenes. The Au‐catalyzed oxidative coupling is driven by the local electrochemical potential at the nanostructured Au STM tip. The products are detected in situ by measuring the conductance and molecular junction elongation and compared with analogous measurements of the expected azobenzene derivatives prepared ex situ. This single‐molecule approach is robust, and it can quickly and reproducibly create reactions for a variety of anilines. We further demonstrate the selective synthesis of geometric isomers and the assembly of complex molecular architectures by sequential coupling of complementary anilines, demonstrating unprecedented control over bond formation at the nanoscale.
Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication.

Huang, H.; Strater, Z. M.; Rauch, M.; Shee, J.; Sisto, T. J.; Nuckolls, C.; Lambert, T. H. Angew. Chem. Int. Ed. 2019, 58, 13318-13322.
Visible‐light photocatalysis and electrocatalysis are two powerful strategies for the promotion of chemical reactions. Here, these two modalities are combined in an electrophotocatalytic oxidation platform. This chemistry employs a trisaminocyclopropenium (TAC) ion catalyst, which is electrochemically oxidized to form a cyclopropenium radical dication intermediate. The radical dication undergoes photoexcitation with visible light to produce an excited‐state species with oxidizing power (3.33 V vs. SCE) sufficient to oxidize benzene and halogenated benzenes via single‐electron transfer (SET), resulting in C−H/N−H coupling with azoles. A rationale for the strongly oxidizing behavior of the photoexcited species is provided, while the stability of the catalyst is rationalized by a particular conformation of the cis‐2,6‐dimethylpiperidine moieties.
Controlled Cationic Polymerization: Single-Component Initiation Under Ambient Conditions.

Kottisch, V.; O’Leary, J. M.; Michaudel, Q.; Stache, E.; Lambert, T. H.; Fors, B. J. Am. Chem. Soc. 2019, 141, 10605-10609.
Cationic polymerizations provide a valuable strategy for preparing macromolecules with excellent control but are inherently sensitive to impurities and commonly require rigorous reagent purification, low temperatures, and strictly anhydrous reaction conditions. By using pentacarbomethoxycyclopentadiene (PCCP) as the single-component initiating organic acid, we found that a diverse library of vinyl ethers can be controllably polymerized under ambient conditions. Additionally, excellent chain-end fidelity is maintained even without rigorous monomer purification. We hypothesize that a tight ion complex between the PCCP anion and the oxocarbenium ion chain end prevents chain-transfer events and enables a polymerization with living characteristics. Furthermore, terminating the polymerization with functional nucleophiles allows for chain-end functionalization in high yields.
Development of a Hydrazine-Catalyzed Carbonyl-Olefin Metathesis Reaction.

Lambert, T. H. Synlett 2019, 30, 1954-1965.
Carbonyl-olefin metathesis is a potentially powerful yet underexplored reaction in organic synthesis. In recent years, however, this situation has begun to change, most notably with the introduction of several different catalytic technologies. The development of one of those new strategies, based on hydrazine catalysts and a novel [3+2] paradigm for double bond metathesis, is discussed herein. First, the stage is set with a description of some potential applications of carbonyl-olefin metathesis and a discussion of alternative strategies for this intriguing reaction.
Oxidizable Ketones: Persistent Radical Cations from the Single Electron Oxidation of 2,3‐Diaminocyclopropenones.

Strater, Z. M.; Rauch, M.; Jockusch, S.; Griffith, A. K.; Lambert, T. H. Angew. Chem. Int. Ed. 2019, 58, 8049-8052.
Single electron oxidation of 2,3‐diaminocyclopropenones is shown to give rise to stable diaminocyclopropenium oxyl (DACO) radical cations. Cyclic voltammetry reveals reversible oxidations in the range of +0.70–1.10 V (vs. SCE). Computational, EPR, and X‐ray analysis support the view that the oxidized species is best described as a cyclopropenium ion with spin density located on the heteroatom substituents, including 23.5 % on oxygen. The metal–ligand behavior of the DACO radical is also described.
A Scalable, One-Pot Synthesis of 1,2,3,4,5-Pentacarbomethoxycyclopentadiene.

Radtke, M. A.; Dudley, C. C.; O'Leary, J. M.; Lambert, T. H. Synthesis 2019, 51, 1135-1138.
1,2,3,4,5-Pentacarbomethoxycyclopentadiene (PCCP) is a strong organic acid and a precursor to useful organocatalysts, including chiral Brønsted acids and silicon-based Lewis acids. The synthetic route to PCCP, first reported in 1942, is inconvenient for a number of reasons. The two-step synthesis requires the purification of intermediates from intractable side-products, high reaction temperatures, and extensive labor (3 days). We have developed an improved procedure that delivers PCCP efficiently in 24 hours in one pot at ambient temperature and without isolation.
The Hydrazine–O2 Redox Couple as a Platform for Organocatalytic Oxidation: Benzo[c]cinnoline-Catalyzed Oxidation of Alkyl Halides to Aldehydes.

Stone, I. B.; Jermaks, J.; MacMillan, S. N.; Lambert, T. H. Angew. Chem. Int. Ed. 2018, 57, 12494-12498.
An organocatalytic oxidation platform that capitalizes on the capacity of hydrazines to undergo rapid autoxidation to diazenes is described. Commercially available benzo[c]cinnoline is shown to catalyze the oxidation of alkyl halides to aldehydes in a novel mechanistic paradigm involving nucleophilic attack, prototropic shift, and hydrolysis. The hydrolysis and reoxidation events occur readily with only adventitious oxygen and water. A survey of the scope of viable substrates is shown along with mechanistic and computational studies that give insight into this mode of catalysis.
Silylated Cyclopentadienes as Competent Silicon Lewis Acid Catalysts.

Radtke, M. A.; Lambert, T. H. Chem. Sci. 2018, 9, 6406–6410.
The synthesis and characterization of silicon Lewis acid complexes that incorporate highly electron-deficient cyclopentadienes is reported. Several pentacarboxycyclopentadienyl and monocarboxytetracyanocyclopentadienyl complexes were prepared. A comparison of their reactivities for catalysis of the allylation of an electron-deficient benzaldehyde was established. The use of a monocarboxytetracyano silylium donor was shown to be effective for the allylation or arylation of a variety of electrophiles via an anion abstraction pathway.
Asymmetric Induction via a Helically Chiral Anion: Enantioselective PCCP Bronsted Acid-Catalyzed Inverse Electron-Demand Diels-Alder Cycloaddition of Oxocarbenium Ions.

Gheewala, C.; Hirschi, J. S.; Lee, W.-H.; Paley, D. W.; Vetticatt, M. J.; Lambert, T. H. J. Am Chem. Soc. 2018, 140, 3523-3527.
An enantioselective catalytic inverse-electron-demand Diels–Alder reaction of salicylaldehyde acetal-derived oxocarbenium ions and vinyl ethers to generate 2,4-dioxychromanes is described. Chiral pentacarboxycyclopentadiene (PCCP) acids are found to be effective for a variety of substrates. Computational and X-ray crystallographic analyses support the unique hypothesis that an anion with point-chirality-induced helical chirality dictates the absolute sense of stereochemistry in this reaction.
Ion Transport in Cyclopropenium-Based Polymerized Ionic Liquids.

Griffin, P. J.; Freyer, J. L.; Han, N.; Yin, X.; Gheewala, C.; Lambert, T. H.; Campos, L. M.; Winey, K. I. Macromolecules 2018, 51, 1681-1687.
Ion transport in polymerized ionic liquids (poly-ILs) occurs via a fundamentally different mechanism than in monomeric ionic liquids, and recently progress has been made toward understanding ion conduction in poly-ILs. To gain insight into the nature of ionic conductivity in ionic polymers, we investigate the physical properties of the trisaminocyclopropenium (TAC) ion, as it is an aromatic carbocation with unique structural and electronic properties. Herein, we characterize the thermal properties, local morphology, and dielectric response of a series of monomeric and polymeric TAC ionic liquids with different counterions. We have found that the extent of a “superionic” mechanism depends on the nature of the ion pair and can result in anomalously high conductivity at the calorimetric Tg. Our results suggest that the molecular volumes of the cationic and anionic species are important parameters that impact ion conductivity in polymerized ionic liquids.
When Size Matters: Exploring the Potential of Aminocyclopropenium Cations as Head Groups in Triphenylene-Derived Ionic Liquid Crystals in Comparison with Guanidinium and Ammonium Units.

Litterscheidt, J.; Judge, P.; Bühlmeyer, A.; Bader, K.; Bandar, J. S.; Lambert, T. H.; Laschat, S. Liquid Crystals 2018, 45, 1250-1258.
The influence of the size of a single ionic head group on the mesomorphic properties of hexaalkoxytriphenylenes was investigated by synthesising three derivatives with increasing head group diameter. The derivatives were investigated with optical polarising microscopy (POM), differential scanning calorimetry (DSC) and X-ray scattering (WAXS, SAXS). For the derivative with the small trimethylammonium head group, an enantiotropic mesophase was found. The derivative with the bigger tetramethylguanidine head group only showed a monotropic phase and the derivative with the largest bisdiisopropylaminocyclopropenium head group displayed no liquid crystaline properties at all.
Cross-coupling of Sulfonic Acid Derivatives via Aryl Radical Transfer (ART) using TTMSS or Photoredox.

Nacsa, E. D.; Lambert, T. H. Org. Chem. Front. 2018, 5, 64-69.
The intramolecular cross-coupling of sulfonic acid derivatives occurs in the presence of tris(trimethylsilyl)silane (TTMSS) at room temperature and in air to form biaryl compounds. A photoredox-catalyzed procedure is also described. These protocols provide mild and convenient alternatives to standard tin-mediated reactions. Combined with the trivial preparation of the substrates from activated sulfonic acids and 2-halophenols or anilines, this work presents a useful means to employ sulfonic acid derivatives in cross-coupling transformations. A modified linker to realize high regioselectivity is also presented. Finally, a one-pot cross-coupling procedure is demonstrated.
Methods for the Synthesis of Functionalized Pentacarboxycyclopentadienes.

Gheewala, C. D.; Radtke, A. M.; Hui, J.; Hon, A. B.; Lambert, T. H. Org. Lett. 2017, 19, 4227-4230.
Protocols for the synthesis of diverse pentacarboxycyclopentadienes are described. Starting from readily available pentacarbomethoxycyclopentadiene, transesterification offers single-step access to aliphatic ester derivatives, while treatment with amines produces mono- or diamides. For less nucleophilic alcohols, an alternative procedure involving the in situ generation and esterification of a putative cyclopentadiene pentaacid chloride has been developed.
Stimulated Raman Scattering of Polymer Nanoparticles for Multiplexed Live-Cell Imaging.

Hu, F.; Brucks, S. D.; Lambert, T. H.; Campos, L. M.; Min, W. Chem. Commun. 2017, 53, 6187-6190.
A novel nanoparticle-based imaging strategy is introduced that couples biocompatible organic nanoparticles and stimulated Raman scattering (SRS) microscopy. Polymer nanoparticles with vibrational labels incorporated were readily prepared for multi-color SRS imaging with excellent photo-stability. The Raman-active polymer dots are nontoxic, rapidly enter various cell types, and are applied in multiplexed cell-type sorting.
Influence of Substituent Chain Branching on the Transfection Efficacy of Cyclopropenium-Based Polymers.

Brucks, S. D.; Freyer, J. L.; Lambert, T. H.; Campos, L. M. Polymers 2017, 9, 79-87.
The realization of gene therapy relies on the development of delivery vectors with high efficiency and biocompatibility. With a multitude of structures accessible, the core challenge is precisely tuning vector structure to probe and optimize structure–property relationships. Employing a modular strategy, two pairs of cationic polymers based on the trisaminocyclopropenium (TAC) ion were synthesized where the substituents differ in the degree of alkyl chain branching. All TAC-based polymers exhibited higher transfection efficiencies than the untreated controls, with variable in vitro toxicities. Considering both cytotoxicity and transfection efficacy, an optimal nonviral vector was identified. Our studies highlight the importance of exercising precise control over polymer structure, both in terms of backbone identity and substituent nature, and the necessity of a robust, modular platform from which to study them.
Clickable Poly(ionic liquids): A Materials Platform for Transfection.

Freyer, J. L.; Brucks, S. D.; Gobieski, G. S.; Russell, S. T.; Yozwiak, C. E.; Sun, M.; Chen. Z.; Jiang, Y.; Bandar, J. S.; Stockwell, B. R.; Lambert, T. H.; Campos, L. M. Angew. Chem. Int. Ed. 2016, 55, 12382-12386.
The potential applications of cationic poly(ionic liquids) range from medicine to energy storage, and the development of efficient synthetic strategies to target innovative cationic building blocks is an important goal. A post‐polymerization click reaction is reported that provides facile access to trisaminocyclopropenium (TAC) ion‐functionalized macromolecules of various architectures, which are the first class of polyelectrolytes that bear a formal charge on carbon. Quantitative conversions of polymers comprising pendant or main‐chain secondary amines were observed for an array of TAC derivatives in three hours using near equimolar quantities of cyclopropenium chlorides. The resulting TAC polymers are biocompatible and efficient transfection agents. This robust, efficient, and orthogonal click reaction of an ionic liquid, which we term ClickabIL, allows straightforward screening of polymeric TAC derivatives. This platform provides a modular route to synthesize and study various properties of novel TAC‐based polymers.
Macrosteres: The Deltic Guanidinium Ion.

Mishiro, K.; Hu, F.; Paley, D. W.; Min, W.; Lambert, T. H. Eur. J. Org. Chem. 2016, 1655-1659.
The “deltic guanidinium” ion is described here as a “macrostere” of the guanidinium ion. The use of the 2,4‐dimethoxybenzyl protecting group allows for the synthesis of the fully unsubstituted parent compound and a variety of derivatives bearing multiple N–H functions for the first time. Deltic urea, deltic thiourea, and deltic benzamidine are also synthesized. A comparison of the physical properties of guanidinium and deltic guanidinium ions is provided. The use of a deltic guanidinium dendrimer for cell transport is demonstrated.
An Aromatic Ion Platform for Enantioselective Brønsted Acid Catalysis.

Gheewala, C. D.; Collins, B. E.; Lambert, T. H. Science 2016, 351 , 961-965.
Chiral acid catalysts are useful for the synthesis of enantioenriched small molecules, but the standard catalysts require laborious and expensive preparations. Here, we describe a chiral Brønsted acid prepared in one step from naturally occurring (–)-menthol and readily available 1,2,3,4,5-pentacarbomethoxycyclopentadiene. Aromatic stabilization serves as a key contributing factor to the potent acidity of the resulting compound, which is shown to catalyze both Mukaiyama-Mannich and oxocarbenium aldol reactions with high efficiency and enantioselectivity. Catalyst loadings as low as 0.01 mole percent and preparative scalability (25 grams) are demonstrated. Alternative amide catalysts are also shown to be promising platforms. In addition to proton catalysis, a chiral anion pathway is demonstrated to be viable with this catalyst system.
Cyclopropenimine Superbases: Competitive Initiation Processes in Lactide Polymerization.

Stukenbroeker, T. S.; Bandar, J. S.; Zhang, X.; Lambert, T. H.; Waymouth, R. M. ACS Macro Lett. 2015, 4, 853-856.
Cyclopropenimine superbases were employed to catalyze the ring-opening polymerization of lactide. Polymerization occurred readily in the presence and absence of alcohol initiators. Polymerizations in the absence of alcohol initiators revealed a competitive initiation mechanism involving deprotonation of lactide by the cyclopropenimine to generate an enolate. NMR and MALDI-TOF analysis of the poly(lactides) generated from cyclopropenimines in the absence of alcohol initiators showed acylated lactide and hydroxyl end groups. Model studies and comparative experiments with guanidine and phosphazene catalysts revealed the subtle influence of the nature of the superbase on competitive initiation processes.
Higher-Order Cyclopropenimine Superbases. Direct Neutral Brønsted Base Catalyzed Michael Reactions with α-Aryl Esters.

Nacsa, E. D.; Lambert, T. H. J. Am. Chem. Soc. 2015, 137, 10246-10253.
The synthesis and characterization of six new classes of higher-order superbases, including five that incorporate cyclopropenimine functionality, has been achieved. We propose a nomenclature that designates these as the CG2, GC2, PC3, PC1, C3, and GP2 classes of superbases. The pKBH+ values were measured to be between 29.0 and 35.6 in acetonitrile. Linear correlations of ten superbase basicities vs that of their substituents demonstrated the insulating effect of the cyclopropenimine core. The molecular structures of several of these materials were obtained by single-crystal X-ray analysis, revealing interesting aspects of conformational bias and noncovalent organization. The types of superbasic cores and substituents were each reliably shown to affect selectivity for deprotonation over alkylation. Higher-order cyclopropenimine and guanidine superbase stability to hydrolysis was found to correlate to basicity. Finally, a GC2 base was found to catalyze conjugate additions of α-aryl ester pronucleophiles, representing the first report of a neutral Brønsted base to catalyze such reactions.
Phase-Transfer and Other Types of Catalysis with Cyclopropenium Ions.

Bandar, J. S.; Tanaset, A.; Lambert, T. H. Chem. Eur. J. 2015, 21 , 7365-7368.
This work establishes the cyclopropenium ion as a viable platform for efficient phase‐transfer catalysis of a diverse range of organic transformations. The amenability of these catalysts to large‐scale synthesis and structural modification is demonstrated. Evaluation of the molecular structure of an optimal catalyst reveals some unique structural features of these systems. Finally, a discussion of electronic charge distribution underscores an important consideration for catalyst design.
The Evolution of Cyclopropenium Ions into Functional Polyelectrolytes.

Jiang, Y.; Freyer, J. L.; Cotanda, P.; Brucks, S. D.; Killops, K. L.; Bandar, J. S.; Torsitano, C.; Balsara, N. P.; Lambert, T. H.; Campos, L. M. Nature Commun. 2015, 6, 5950.
Versatile polyelectrolytes with tunable physical properties have the potential to be transformative in applications such as energy storage, fuel cells and various electronic devices. Among the types of materials available for these applications, nanostructured cationic block copolyelectrolytes offer mechanical integrity and well-defined conducting paths for ionic transport. To date, most cationic polyelectrolytes bear charge formally localized on heteroatoms and lack broad modularity to tune their physical properties. To overcome these challenges, we describe herein the development of a new class of functional polyelectrolytes based on the aromatic cyclopropenium ion. We demonstrate the facile synthesis of a series of polymers and nanoparticles based on monomeric cyclopropenium building blocks incorporating various functional groups that affect physical properties. The materials exhibit high ionic conductivity and thermal stability due to the nature of the cationic moieties, thus rendering this class of new materials as an attractive alternative to develop ion-conducting membranes.
Structure-Activity Relationship Studies of Cyclopropenimines as Enantioselective Bronsted Base Catalysts.

Bandar, J. S.; Barthelme, A. P.; Mazori, A. Y.; Lambert, T. H. Chem. Sci. 2015, 6, 1537-1547.
We recently demonstrated that chiral cyclopropenimines are viable Brønsted base catalysts in enantioselective Michael and Mannich reactions. Herein, we describe a series of structure–activity relationship studies that provide an enhanced understanding of the effectiveness of certain cyclopropenimines as enantioselective Brønsted base catalysts. These studies underscore the crucial importance of dicyclohexylamino substituents in mediating both reaction rate and enantioselectivity. In addition, an unusual catalyst CH⋯O interaction, which provides both ground state and transition state organization, is discussed. Cyclopropenimine stability studies have led to the identification of new catalysts with greatly improved stability. Finally, additional demonstrations of substrate scope and current limitations are provided herein.
2,3-Diazabicyclo[2.2.1]heptane.
Transition State Analysis of Enantioselective Brønsted Base Catalysis by Chiral Cyclopropenimines.

Bandar, J. S.; Sauer, G. S.; Wulff, W. D.; Lambert, T. H.; Vetticatt, M. J. J. Am. Chem. Soc. 2014, 136, 10700-10707.
Experimental 13C kinetic isotope effects have been used to interrogate the rate-limiting step of the Michael addition of glycinate imines to benzyl acrylate catalyzed by a chiral 2,3-bis(dicyclohexylamino) cyclopropenimine catalyst. The reaction is found to proceed via rate-limiting carbon–carbon bond formation. The origins of enantioselectivity and a key noncovalent CH···O interaction responsible for transition state organization are identified on the basis of density functional theory calculations and probed using experimental labeling studies. The resulting high-resolution experimental picture of the enantioselectivity-determining transition state is expected to guide new catalyst design and reaction development.
Synthesis and Characterization of a Diaziridinium Ion. Conversion of 3,4-Dihydroisoquinolines to 4,5-Dihydro-3H-benzo[2,3]diazepines via a formal N-Insertion Process.

Allen, J. M.; Lambert, T. H. Tetrahedron 2014, 70, 4111-4117.
A diaziridinium ion has been synthesized in high yield and its structure unambiguously confirmed by X-ray crystal analysis. The predicted N-transfer reactivity with olefins of this species was not observed. Instead, upon heating, the diaziridinium ion underwent ring opening to produce a dihydrobenzodiazepene product in good yield, thus achieving a formal N-insertion of the starting dihydroisoquinoline substrate. This process has been demonstrated on 11 total substrates.
The Development of Catalytic Nucleophilic Substitution Reactions: Challenges, Progress, and Future Directions.

An, J.; Denton, R. M.; Lambert, T. H.; Nacsa, E. Org. Biomol. Chem. 2014, 12, 2993.
Bimolecular nucleophilic substitution reactions of alcohols are fundamentally important transformations in organic chemistry yet, to date, they are relatively underdeveloped with respect to catalysis. This Article describes the emerging area of catalytic SN2 reactions with specific emphasis on the design and development of phosphorus(V) and cyclopropenone-based catalytic SN2 reactions of alcohols.
Distortion-Accelerated Cycloadditions and Strain-Release-Promoted Cycloreversions in the Organocatalytic Carbonyl-Olefin Metathesis.

Hong, X.; Liang, Y.; Griffith, A. K.; Lambert, T. H.; Houk, K. N. Chem. Sci. 2014, 5, 471-475.
The mechanism of hydrazine-catalyzed carbonyl-olefin metathesis relying on a novel (3 + 2) strategy is studied by density functional theory (DFT) calculations. The origins of the special reactivity of cyclopropene in this transformation are revealed, and the reactivities of different alkenes in the (3 + 2) cycloadditions and cycloreversions are compared. It is found that the ease of distortion of reactants accelerates cycloadditions, and that the strain release is the controlling factor for cycloreversions.
Cyclopropenimine-Catalyzed Enantioselective Mannich Reactions of t-Butyl Glycinates with N-Boc-Imines.

Bandar, J. S.; Lambert, T. H. J. Am. Chem. Soc. 2013, 135, 11799-11802.
Cyclopropenimine 1 is shown to catalyze Mannich reactions between glycine imines and N-Boc-aldimines with high levels of enantio- and diastereocontrol. The reactivity of 1 is shown to be substantially greater than that of a widely used thiourea cinchona alkaloid-derived catalyst. A variety of aryl and aliphatic N-Boc-aldimines are effective substrates for this transformation. A preparative-scale reaction to deliver >90 mmol of product is shown using 1 mol % catalyst. The products of this transformation can be converted into several useful derivatives.
Aminocyclopropenium Ions: Synthesis, Properties, and Applications.

Bandar, J. S.; Lambert, T. H. Synthesis 2013, 45, 2485-2498.
This review covers the preparation, physical properties, and applications of cyclopropenium ions bearing one, two, or three amino substituents. It provides a description of the most reliable methods to access these unique structures as well as a discussion of the reactivity profiles of the common subtypes.
Cyclopropenone Catalyzed Substitution of Alcohols with Mesylate Ion.

Nacsa, E. D.; Lambert, T. H. Org. Lett. 2013, 15, 38-41.
The cyclopropenone catalyzed nucleophilic substitution of alcohols by methanesulfonate ion with inversion of configuration is described. This work provides an alternative to the Mitsunobu reaction that avoids the use of azodicarboxylates and generation of hydrazine and phosphine oxide byproducts. This transformation is shown to be compatible with a range of functionality. A cyclopropenone scavenge strategy is demonstrated to aid purification.
Diphenylcyclopropenone.
Organocatalytic Carbonyl-Olefin Metathesis.

Griffith, A. K.; Vanos, C. M.; Lambert, T. H. J. Am. Chem. Soc. 2012, 134, 18581-18584.
The development of a catalytic carbonyl-olefin metathesis strategy is reported, in the context of the ring-opening metathesis of cyclopropenes with aldehydes using a simple hydrazine catalyst. The key to this reaction is a conceptual blueprint for metathesis chemistry that forgoes the traditional reliance on [2 + 2] cycloaddition modes in favor of a [3 + 2] paradigm.
Enantioselective Brønsted Base Catalysis with Chiral Cyclopropenimines.

Bandar, J. S.; Lambert, T. H. J. Am. Chem. Soc. 2012, 134, 5552-5555.
Cyclopropenimines are shown to be a highly effective new class of enantioselective Brønsted base catalysts. A chiral 2,3-bis(dialkylamino)cyclopropenimine catalyzes the rapid Michael reaction of a glycine imine substrate with high levels of enantioselectivity. A preparative scale reaction to deliver 25 g of product is demonstrated, and a trivial large scale synthesis of the optimal catalyst is shown. In addition, the basicity of a 2,3-bis(dialkylamino)cyclopropenimine is measured for the first time and shown to be approximately equivalent to the P1-tBu phosphazene base. An X-ray crystal structure of the protonated catalyst is shown along with a proposed mechanistic and stereochemical rationale.
Development of a Catalytic Platform for Nucleophilic Substitution: Cyclopropenone Catalyzed Chlorodehydration of Alcohols.

Vanos, C. M.; Lambert, T. H. Angew. Chem. Int. Ed. 2011, 50, 12222-12226.
2,3‐Bis‐(p‐methoxyphenyl)cyclopropenone is a highly efficient catalyst for the chlorodehydration of 20 diverse alcohol substrates. With oxalyl chloride as catalytic activator, this nucleophilic substitution proceeded through cyclopropenium‐activated intermediates and resulted in complete stereochemical inversion in substrates with chiral centers.
Demonstration of the Facile Reversibility of Fulvene Formation.

Bandar, J. S.; Coscia, R. W.; Lambert, T. H. Tetrahedron 2011, 67, 4364-4370.
Cleavage of fulvenes under mild conditions and interchange between electron-deficient fulvenes and their constituent cyclopentadienes and imines is demonstrated for the first time. A series of cyclopentadienes and imines are investigated to probe the dependence of fulvene equilibration on structure. The exchange of one fulvene for another is demonstrated in the first reported example of transfulvenation. Finally, the metathesis of two fulvenes to generate all four possible cross products is shown.
Cyclopropenium-Activated Cyclodehydration of Diols.

Kelly, B. D.; Lambert, T. H. Org. Lett. 2011, 13, 740-743.
The dehydrative cyclization of diols to cyclic ethers via cyclopropenium activation is described. Using 2,3-diphenylcyclopropene and methanesulfonic anhydride, a series of 1,4- and 1,5-diols are rapidly cyclized to furnish tetrahydrofurans and tetrahydropyrans in high yield. Eleven total substrates are shown, including a gram scale cyclization of a diterpene derivative.
Tropylium Ion Mediated α-Cyanation of Amines.

Allen, J. M.; Lambert, T. H. J. Am. Chem. Soc. 2011, 133, 1260-1262.
Tropylium ion mediated α-cyanation of amines is described. Even in the presence of KCN, tropylium ion is capable of oxidizing various amine substrates, and the resulting iminium ions undergo salt metathesis with cyanide ion to produce aminonitriles. The byproducts of this transformation are simply cycloheptatriene, a volatile hydrocarbon, and water-soluble potassium tetrafluoroborate. Thirteen total substrates are shown for the α-cyanation procedure, including a gram scale synthesis of 17β-cyanosparteine. In addition, a tropylium ion mediated oxidative aza-Cope rearrangement is demonstrated.
Cyclopropenium-Activated Beckmann Rearrangement. Catalysis Versus Self-Propagation in Reported Organocatalytic Beckmann Rearrangements.
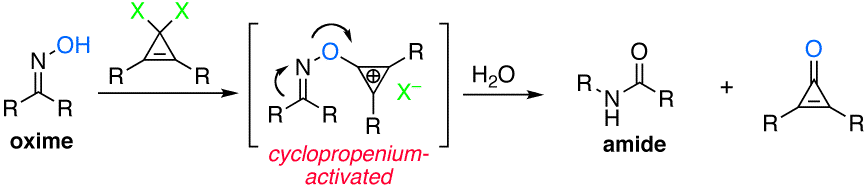
Vanos, C. M.; Lambert, T. H. Chem. Sci. 2010, 1, 705-708.
The concept of cyclopropenium activation has been extended to include dehydrative rearrangements in the context of the Beckmann rearrangement. Geminal dichlorocyclopropenes are shown to rapidly and efficiently convert oximes to amides at room temperature, with reactivity that far surpasses other organic-based promoters. Twelve total examples are provided, including a complex steroidal substrate on preparative scale. Evidence is provided that suggests previously reported organocatalytic Beckmann rearrangements may in fact be self-propagating rather than catalytic.
Multicatalysis: Advancing Synthetic Efficiency and Inspiring Discovery.

Ambrosini, L. M.; Lambert, T. H. Chem. Cat. Chem. 2010, 2, 1373-1380.
Heavy reliance on iterative synthetic strategies represents one of the most serious drawbacks of modern organic synthesis. There has been increasing interest of late in the development of processes that attempt to alleviate this dependence by achieving multiple transformations in a single vessel, thereby circumventing the need for intermittent isolation and purification. One such strategy is encompassed by the term multicatalysis, an approach wherein multiple catalytic reactions are executed in a single flask, either in tandem or sequentially. In this Minireview, some recent efforts in the field of multicatalysis, including our own work, are discussed. In addition, a case regarding the need for consistent terminology in this rapidly developing field is advanced.
Nucleophilic Acyl Subsitution via Aromatic Cation Activation of Carboxylic Acids: Rapid Generation of Acid Chlorides Under Mild Conditions.

Hardee, D. J.; Kovalchuke, L.; Lambert, T. H. J. Am. Chem. Soc. 2010, 132, 5002-5003.
The first example of aromatic cation-activated nucleophilic acyl substitution has been achieved. The conversion of carboxylic acids to their corresponding acid chlorides occurs rapidly in the presence of 3,3-dichlorocyclopropenes via the intermediacy of cyclopropenium carboxylate complexes. The effect of cyclopropene substituents on the rate of conversion is examined. The addition of tertiary amine base is found to dramatically accelerate reaction, and conditions were developed for the preparation of acid sensitive acid chlorides. Preparative scale peptide couplings of two N-Boc amino acids were achieved with this method.
Total Synthesis of the Tylophora Alkaloids Rusplinone, 13aa-Secoantofine, and Antofine using a Multicatalytic Oxidative Aminochlorocarbonylation / Friedel-Crafts Reaction.

Ambrosini, L. A.; Cernak, T. A.; Lambert, T. H. Tetrahedron 2010, 66, 4882-4887.
A rapid synthetic approach to the tylophora alkaloids antofine and 13aα-secoantofine is presented that makes use of a multicatalytic oxidative aminochlorocarbonylation/Friedel–Crafts reaction as the key step. This reaction, along with a one-pot, three-step telescoped process offers a three or four-pot sequence to access the title compounds in high overall yield.
Development of Oxidative Formylation and Ketonylation Reactions.

Ambrosini, L. A.; Cernak, T. A.; Lambert, T. H. Synthesis - Featured Article 2010, 870-881.
The first oxidative formylation and oxidative ketonylation of alkenylamines and alkenyl alcohols is demonstrated. A range of substrates that participate in this process are provided. Oxidative formylation was found to proceed optimally with the use of triphenylsilane as the hydride source. Oxidative ketonylation was feasible with a number of organometallic partners, especially dialkylzinc or organostannanes. An interesting finding regarding the fate of various acylpalladium intermediates is discussed.
Aromatic Cation Activation of Alcohols: Conversion to Alkyl Chlorides Using Dichlorodiphenylcyclopropene.

Kelly, B. D.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 13930-13931.
A novel paradigm for the activation of alcohols toward nucleophilic displacement via formation of cyclopropenium ethers is described. The conversion of a range of alcohol substrates to the corresponding alkyl chlorides occurs rapidly upon treatment with 3,3-dichloro-1,2-diphenylcyclopropene. 1H NMR data support the intermediacy of a cyclopropenium intermediate, and the reaction is demonstrated to proceed primarily via the SN2 mechanism for 1-phenylethanol. A total of 12 examples of substrate scope are provided.
Leaving Group Potential of a Substituted Cyclopentadienyl Anion Towards Oxidative Addition.

Fisher, E. L.; Lambert, T. H. Org. Lett. 2009, 11, 4108-4110.
The facility with which a substituted cyclopentadienyl anion may function as a leaving group for palladium-catalyzed allylation reactions is demonstrated. Reaction of several allylcyclopentadienyl substrates is shown. Nucleophilic displacement of carbon with nitrogen is achieved in the deallylation of allylpenta-p-acetylphenylcyclopentadiene with N-methylbenzylamine.
Lanthanum(III) Triflate-Catalyzed Cyclopropanation via Intramolecular Methylene Transfer.

Hardee, D. J.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 7536-7537.
A new method for cyclopropanation involving intramolecular methylene transfer from an epoxide to an olefin has been developed. This La(OTf)3-catalyzed process proceeds with good efficiency and with high stereoselectivity. A range of examples illustrating substrate scope are given along with a mechanistic rationale. Also demonstrated is an asymmetric cyclopropane synthesis that combines enantioselective epoxidation with this methylene-transfer protocol.
Multicatalytic Synthesis of Complex Tetrahydrofurans Involving Bismuth(III) Triflate-Catalyzed Intramolecular Hydroalkoxylation of Unactivated Olefins.

Kelly, B. D.; Allen, J. M.; Tundel, R. E.; Lambert, T. H. Org. Lett. 2009, 11, 1381-1383.
A multicatalytic synthesis of complex tetrahydrofurans has been developed involving a Bi(OTf)3-catalyzed nucleophilic addition/hydroalkoxylation sequence. Complex tetrahydrofuranyl products may be formed rapidly in high yield and with good diastereoselectivity. The demonstrated scope of hydroalkoxylation has also been expanded to include substrates bearing useful functional handles including carboxylate ester, olefin, nitrile, and nitro groups.
Multicatalytic Synthesis of a-Pyrrolidinyl Ketones via a Tandem Palladium(II)/In(III)-Catalyzed Aminochlorocarbonylation/Friedel-Crafts Acylation Reaction.
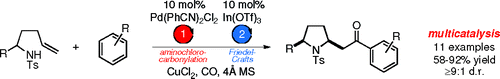
Cernak, T. A.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 3124-3125.
An oxidative carbonylation reaction that generates acid chloride functionality has been developed. Furthermore, this aminochlorocarbonylation reaction has been merged with a catalytic Friedel−Crafts acylation to produce a highly efficient tandem multicatalytic synthesis of pyrrolidinyl ketones. Significant variation of the aromatic nucleophile and substrate are shown. Two examples of incorporation of this method in triple-catalytic sequences are also demonstrated.
Development of a Formal [4+1] Cycloaddition: Pd(OAc)2-Catalyzed Intramolecular Cyclopropanation of 1,3-Dienyl ß-Keto Esters and MgI2-Promoted Vinylcyclopropane-Cyclopentene Rearrangement.

Coscia, R. W.; Lambert, T. H. J. Am. Chem. Soc. 2009, 131, 2496-2498.
A formal [4 + 1] cycloaddition of 1,3-dienyl β-keto esters has been developed. This two step process involves Pd(II)-catalyzed intramolecular cyclopropanation to produce vinylcyclopropanes and a subsequent mild vinylcyclopropane−cyclopentene rearrangement promoted by MgI2. The cyclopropanation method notably requires the use of Mg(ClO4)2, presumably to facilitate keto−enol tautomerization, and is greatly improved by the use of copper(II) isobutyrate as co-oxidant. A range of substrates with various substitution patterns is demonstrated.